四川大学化学学院苏静教授课题组,联合美国洛斯阿拉莫斯国家实验室锕系化学实验和理论研究团队,在锕系元素锎(Cf)的金属有机化学研究方向取得重要突破,以共同第一作者在《Nature》杂志上在线发表了最新研究成果“Isolation and characterization of a californium metallocene”。
锎(Cf)是目前微克级别上制得的最重元素。Cf同位素的稀缺性和严重的放射性危害使得相关实验研究极具挑战性。因此,与其他f元素相比,Cf的5f/6d价轨道参与化学成键的能力、自旋-轨道耦合在电子结构中的作用以及反应类型等一系列化学性质仍处于未知状态。金属有机化合物对解释金属元素的周期性和成键趋势有重要作用。近年来,研究人员对锕系元素钍(Th)至钚(Pu)以及镅(Am) 的金属有机化学性质进行了探索,但鲜有关于锔(Cm)至锎( Cf)的研究报道。
该研究首次合成并表征了一种二茂型锎配合物[Cf(C5Me4H)2Cl2K(OEt2)]n (图1),特别是对Cf元素的化学成键和光谱性质进行了深入的理论研究和计算分析。通过分子轨道能级和成分、Mulliken布居、键级以及QTAIM分析等多角度,解析了锎金属与配体间的成键作用,揭示了Cf-C键以离子键特征为主,共价成分很少(图1)。与镧系类似物[Dy(C5Me4H)2Cl2K(OEt2)]n中Dy 4f轨道相比,Cf 5f轨道对金属与配体间的成键贡献更大,体现在Cf 5f轨道与配体价轨道间的混合程度增加。采用高精度从头算多参考理论方法CASSCF/NEVPT2并包括自旋-轨道耦合效应,精确模拟了锎化合物的UV-vis-NIR光谱,理论预测的光谱特征与实验结果高度一致(图2)。
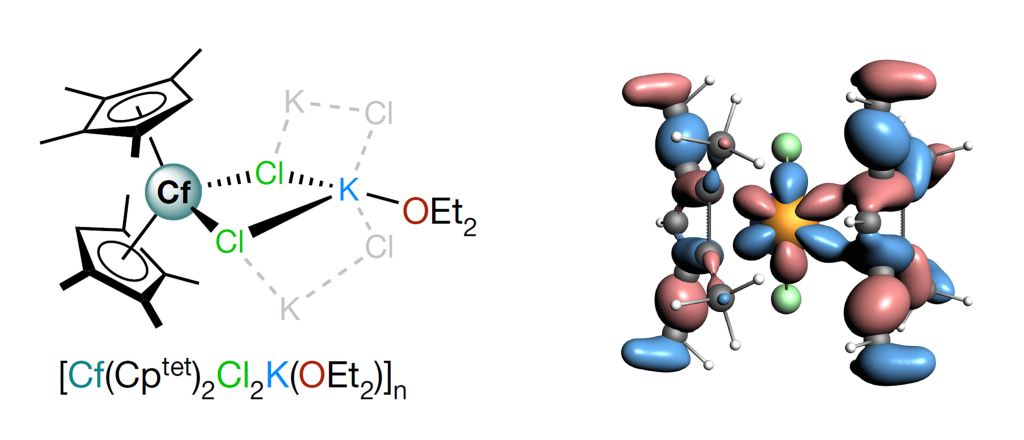
图1. [Cf(C5Me4H)2Cl2K(OEt2)]n的几何结构以及含Cf 5f成分的成键轨道
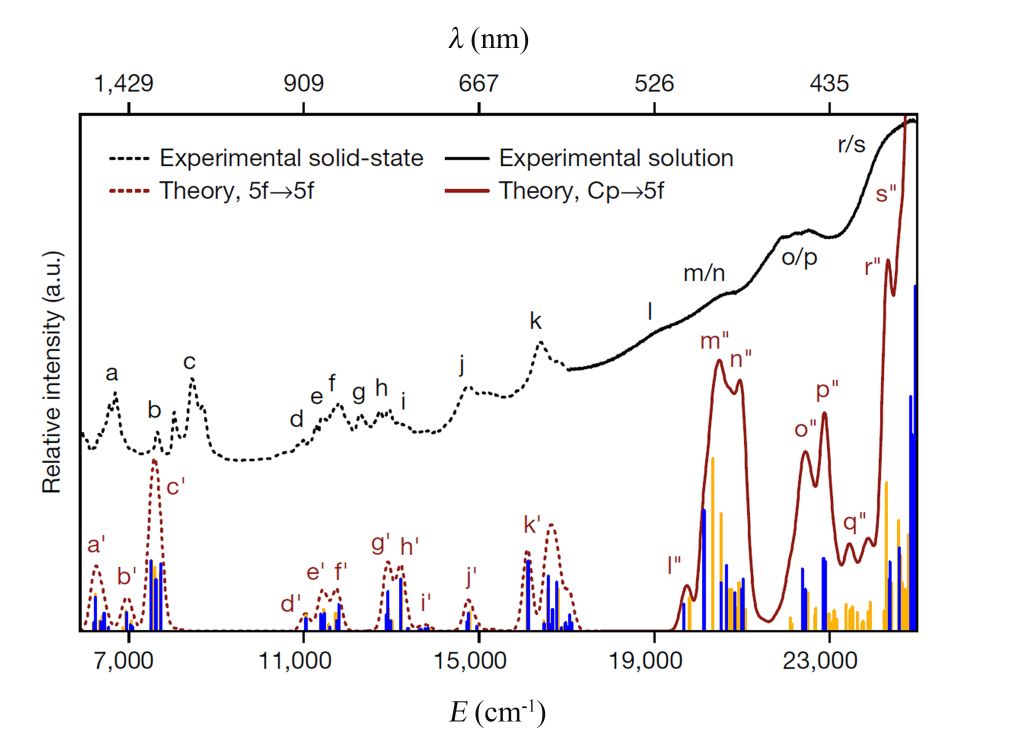
图2. 实验观测与理论模拟的[Cf(C5Me4H)2Cl2K(OEt2)]n化合物的UV-vis-NIR光谱
含时密度泛函理论(TDDFT)计算和分子轨道能级分析很好地解释了深橙色化合物[Cf(C5Me4H)2Cl2K(OEt2)]n与无色化合物[Dy(C5Me4H)2Cl2K(OEt2)]n在颜色方面的差异性。[Cf(C5Me4H)2Cl2K(OEt2)]n中配体价轨道与Cf 5f空轨道的能隙更小,配体到金属的电荷转移跃迁主要发生在可见光区域;而对于[Dy(C5Me4H)2Cl2K(OEt2)]而言,由于Dy 4f空轨道能级较高,这一跃迁则发生在紫外区,因而两者呈现不同的颜色。
理解锕系重元素的物理化学性质是实现核燃料循环中镧锕分离的核心所在,其中理论计算研究起着举足轻重的作用,其为阐释镧锕体系复杂的电子结构和化学成键性质,特别是精确解析实验光谱提供了有力支撑和科学指证。
特别感谢国家自然科学基金委和四川大学对本课题研究支持。
全文链接:https://www.nature.com/articles/s41586-021-04027-8
